Research Spotlight
AMY ROSENZWEIG LAB
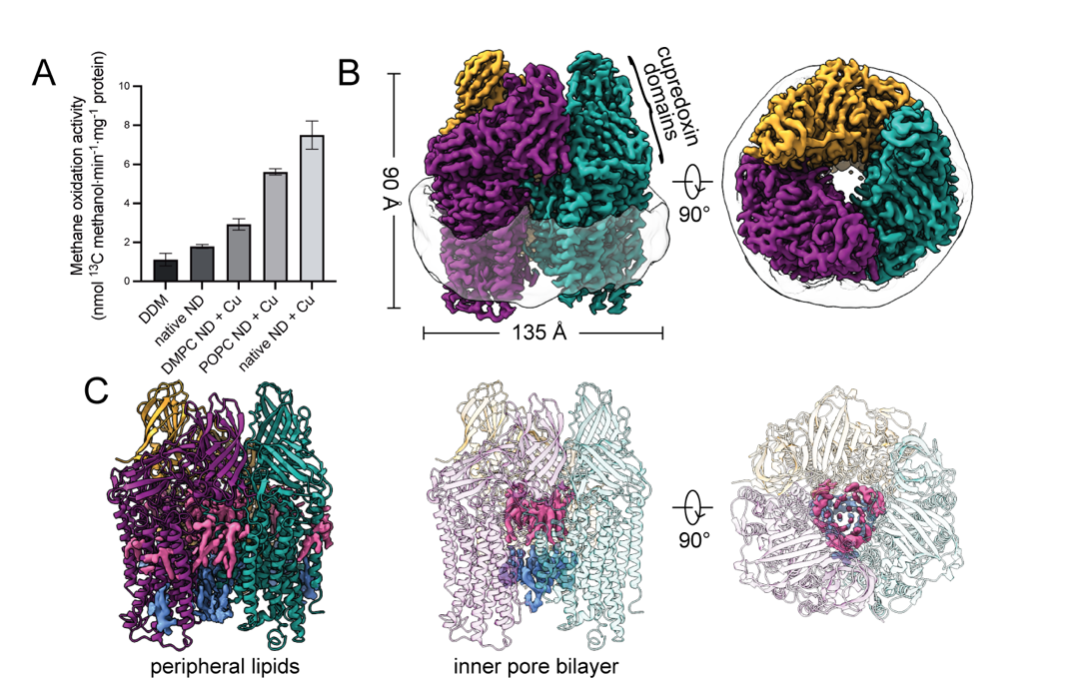
Recovery of particulate methane monooxygenase structure and activity in a lipid bilayer
Christopher W. Koo, Frank J. Tucci, Yuan He, Amy C. Rosenzweig
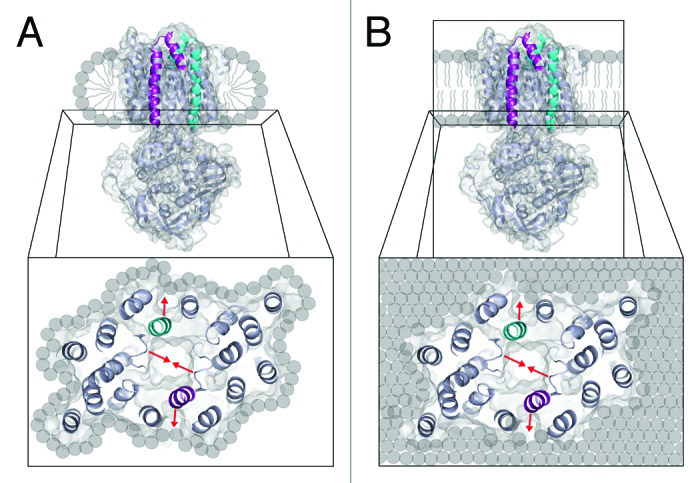
Bacterial methane oxidation using the enzyme particulate methane monooxygenase (pMMO) contributes to removal of environmental methane, a potent greenhouse gas. Crystal structures determined using inactive, detergent-solubilized pMMO lack several highly conserved regions neighboring the proposed active site. We show that reconstituting pMMO in nanodiscs with lipids extracted from the native organism restores methane oxidation activity. Multiple nanodisc-embedded pMMO structures determined by cryoelectron microscopy to 2.14-2.46 Å resolution reveal the structure of pMMO in a lipid environment. The resulting model includes stabilizing lipids, regions of the PmoA and PmoC subunits not observed in prior structures, and a previously undetected copper binding site in the PmoC subunit with an adjacent hydrophobic cavity. These structures provide a revised framework for understanding and engineering pMMO function.
Yuan He Lab
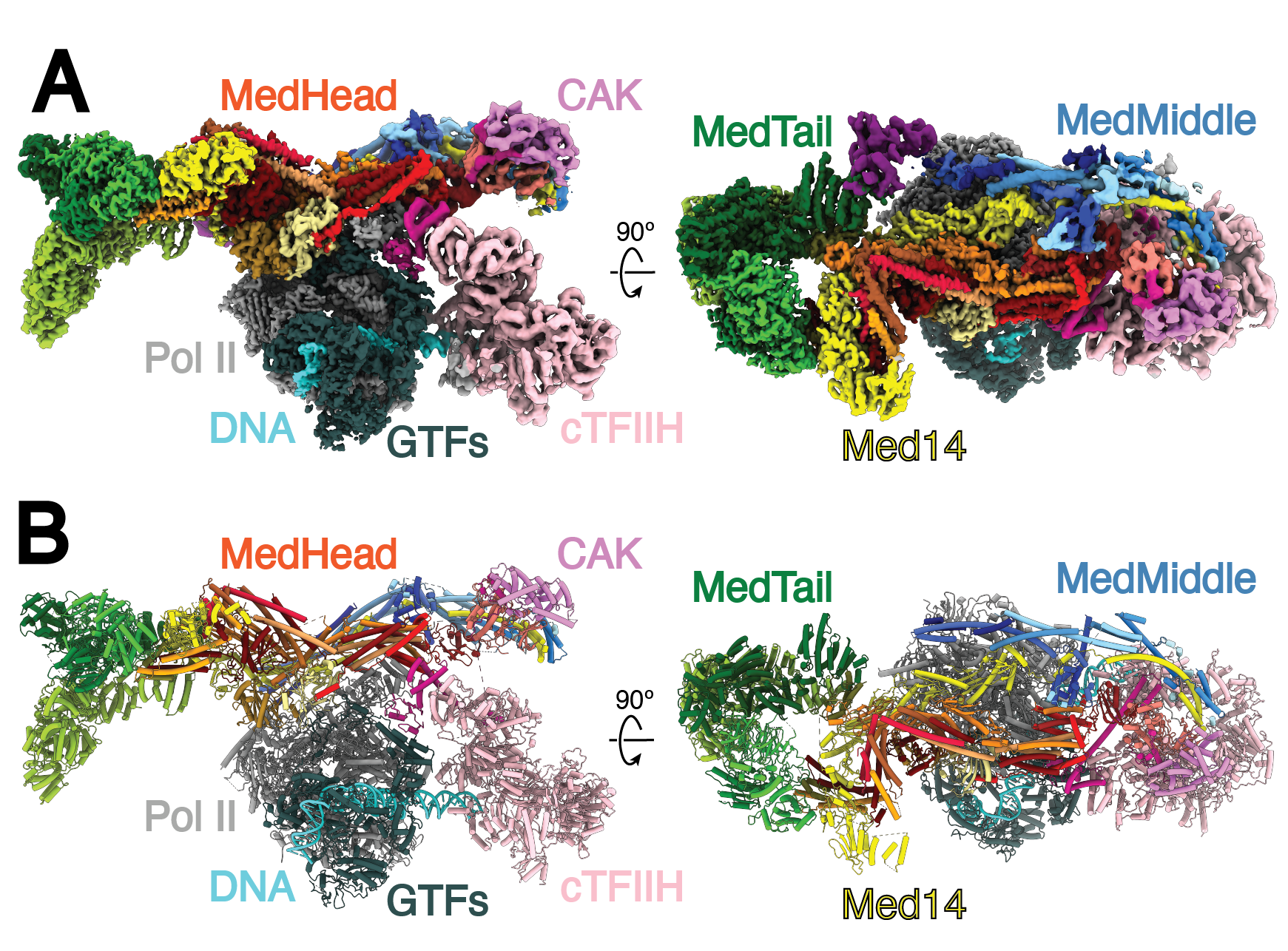
Structure of the human Mediator-bound transcription preinitiation complex
Ryan Abdella, Anna Talyzina, Siyu Chen, C. J. Inouye, R. Tjian, and Yuan He.
Eukaryotic transcription requires the assembly of a multi-subunit preinitiation complex (PIC) comprised of RNA polymerase II (Pol II) and the general transcription factors. The co-activator Mediator is recruited by transcription factors, facilitates the assembly of the PIC, and stimulates phosphorylation of the Pol II C-terminal domain (CTD) by the TFIIH subunit CDK7. Here, we present the cryo-electron microscopy structure of the human Mediator-bound PIC at sub-4 Å. Transcription factor binding sites within Mediator are primarily flexibly tethered to the tail module. CDK7 is stabilized by multiple contacts with Mediator. Two binding sites exist for the Pol II CTD, one between the head and middle modules of Mediator and the other in the active site of CDK7, providing structural evidence for Pol II CTD phosphorylation within the Mediator-bound PIC.
JULIUS LUCKS LAB
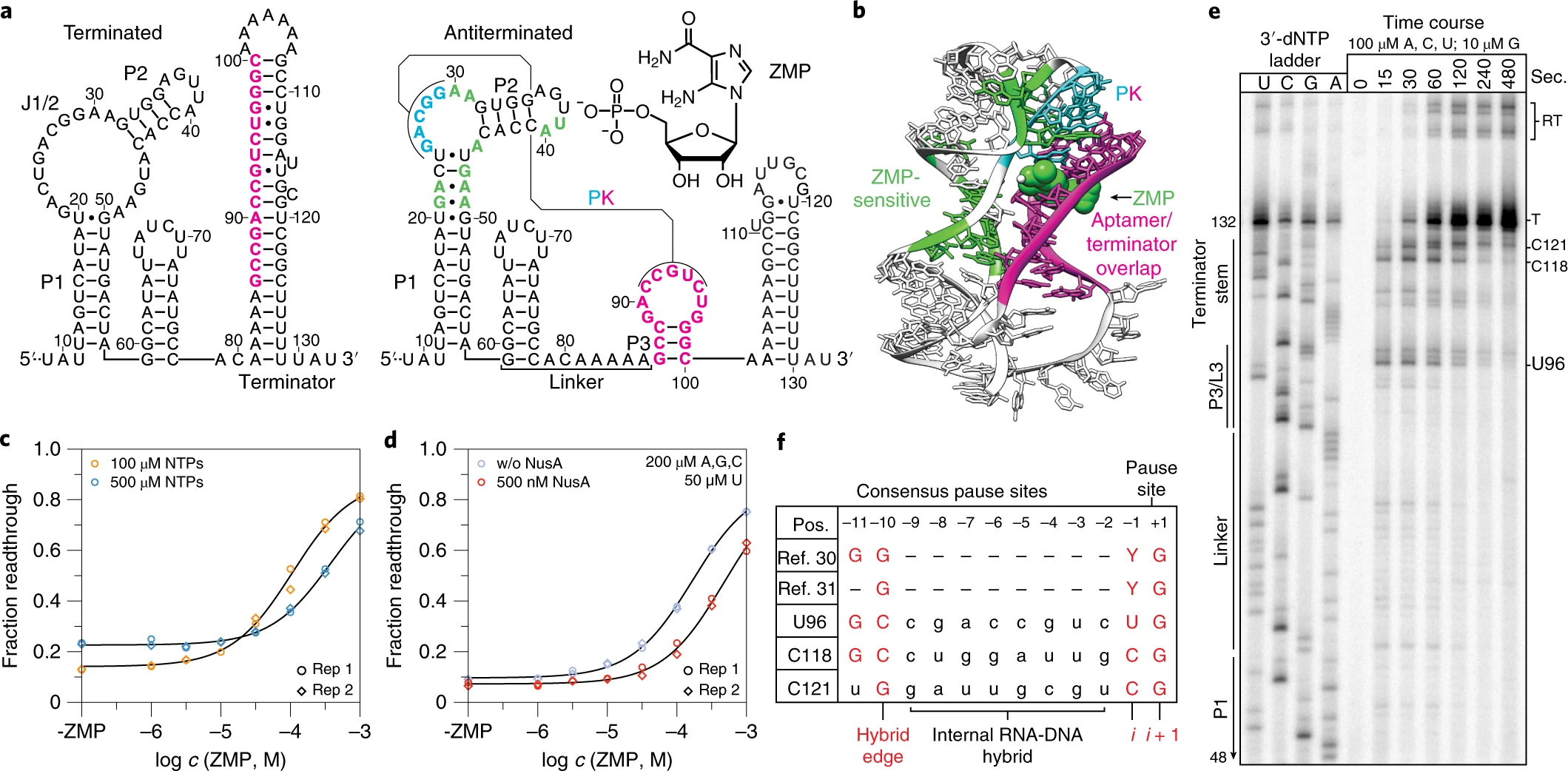
A ligand-gated strand displacement mechanism for ZTP riboswitch transcription control
Eric J. Strobel, Luyi Cheng, Katherine E. Berman, Paul D. Carlson and Julius B. Lucks.
Cotranscriptional folding is an obligate step of RNA biogenesis that can guide RNA structure formation and function through transient intermediate folds. This process is particularly important for transcriptional riboswitches in which the formation of ligand-dependent structures during transcription regulates downstream gene expression. However, the intermediate structures that comprise cotranscriptional RNA folding pathways, and the mechanisms that enable transit between them, remain largely unknown. Here, we determine the series of cotranscriptional folds and rearrangements that mediate antitermination by the Clostridium beijerinckii pfl ZTP riboswitch in response to the purine biosynthetic intermediate ZMP. We uncover sequence and structural determinants that modulate an internal RNA strand displacement process and identify biases within natural ZTP riboswitch sequences that promote on-pathway folding. Our findings establish a mechanism for pfl riboswitch antitermination and suggest general strategies by which nascent RNA molecules navigate cotranscriptional folding pathways.
AMY ROSENZWeIG LAB
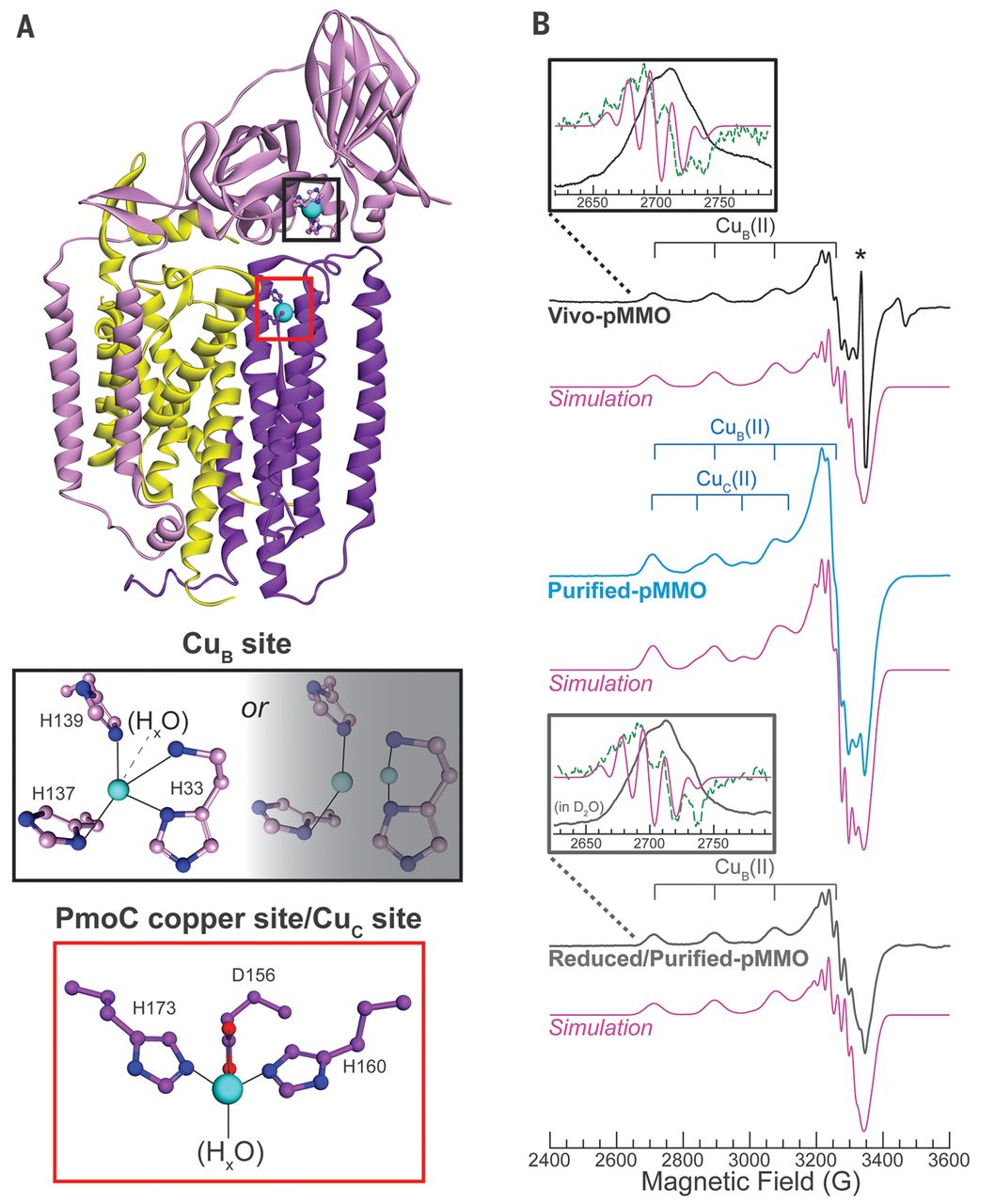
Particulate methane monooxygenase contains only mononuclear copper centers
Matthew O. Ross, Fraser MacMillan, Jingzhou Wang, Alex Nisthal, Thomas J. Lawton, Barry D. Olafson, Stephen L. Mayo, Amy C. Rosenzweig, Brian M. Hoffman
Bacteria that oxidize methane to methanol are central to mitigating emissions of methane, a potent greenhouse gas. The nature of the copper active site in the primary metabolic enzyme of these bacteria, particulate methane monooxygenase (pMMO), has been controversial owing to seemingly contradictory biochemical, spectroscopic, and crystallographic results. We present biochemical and electron paramagnetic resonance spectroscopic characterization most consistent with two monocopper sites within pMMO: one in the soluble PmoB subunit at the previously assigned active site (CuB) and one ~2 nanometers away in the membrane-bound PmoC subunit (CuC). On the basis of these results, we propose that a monocopper site is able to catalyze methane oxidation in pMMO.
NEIL KELLEHER LAB
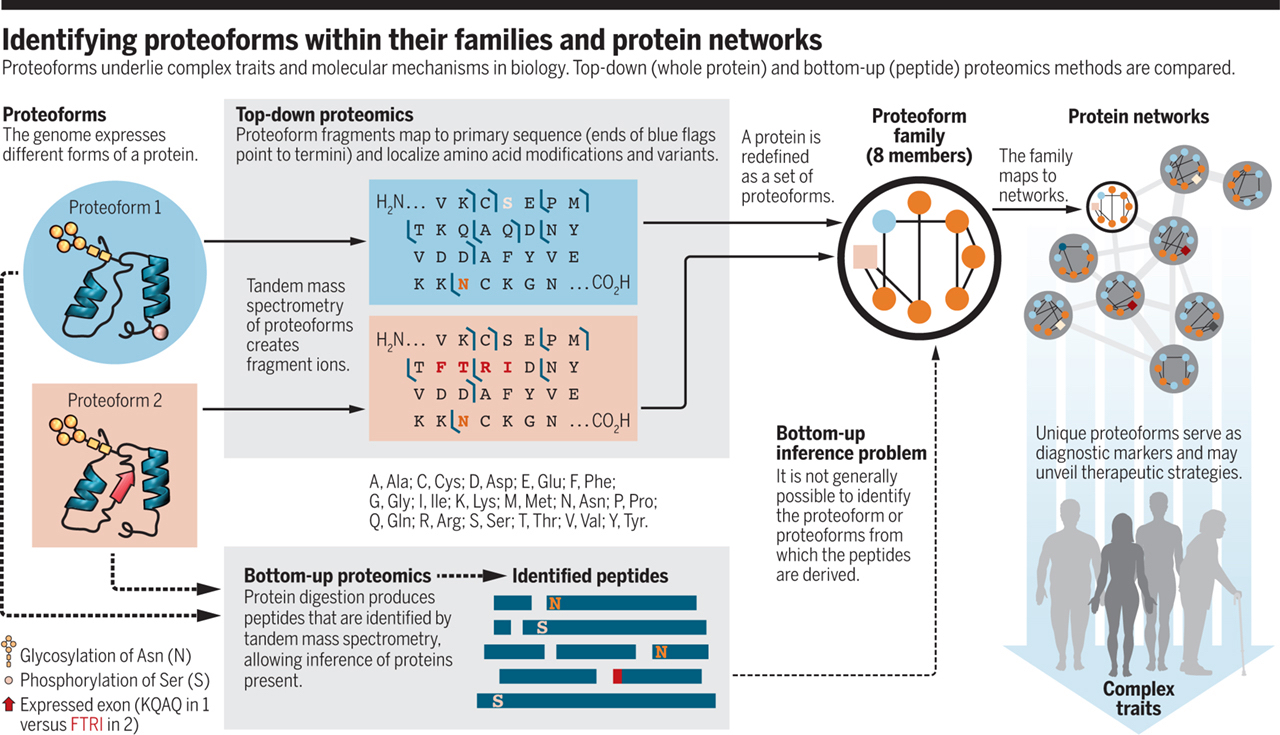
Proteoforms as the next proteomics currency
Lloyd M. Smith and Neil L Kelleher
Proteoforms—the different forms of proteins produced from the genome with a variety of sequence variations, splice isoforms, and myriad posttranslational modifications (1)—are critical elements in all biological systems (see the figure, left). Yang et al. (2) recently showed that the functions of proteins produced from splice variants from a given gene—different proteoforms—can be as different as those for proteins encoded by entirely different genes. Li et al. (3) showed that splice variants play a central role in modulating complex traits. However, the standard paradigm of proteomic analysis, the “bottom-up” strategy pioneered by Eng and Yates some 20 years ago (4), does not directly identify proteoforms. We argue that proteomic analysis needs to provide the identities and abundances of the proteoforms themselves, rather than just their peptide surrogates. Developing new proteome-wide strategies to accomplish this goal presents a formidable but not insurmountable technological challenge that will benefit the biomedical community.
AMY ROSENZWeIG LAB
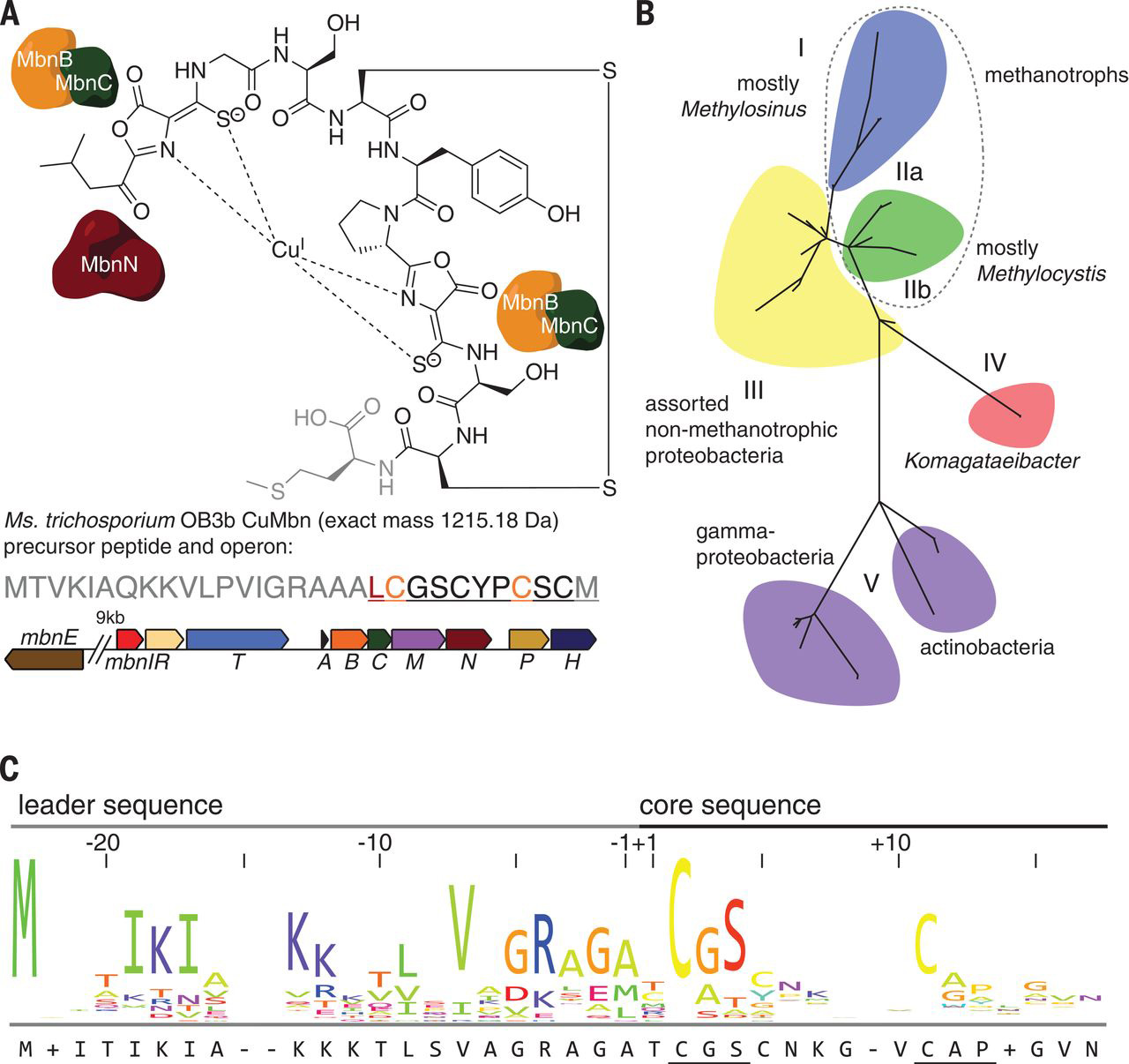
The biosynthesis of methanobactin
Grace E. Kenney, Laura M. K. Dassama, Maria-Eirini Pandelia, Anthony S. Gizzi, Ryan J. Martinie, Peng Gao, Caroline J. DeHart, Luis F. Schachner, Owen S. Skinner, Soo Y. Ro, Xiao Zhu, Monica Sadek, Paul M. Thomas, Steven C Almo, J. Martin Bollinger Jr., Carsten Krebs, Neil L. Kelleher, Amy C. Rosenzweig
Metal homeostasis poses a major challenge to microbes, which must acquire scarce elements for core metabolic processes. Methanobactin, an extensively modified copper-chelating peptide, was one of the earliest natural products shown to enable microbial acquisition of a metal other than iron. We describe the core biosynthetic machinery responsible for the characteristic posttranslational modifications that grant methanobactin its specificity and affinity for copper. A heterodimer comprising MbnB, a DUF692 family iron enzyme, and MbnC, a protein from a previously unknown family, performs a dioxygen-dependent four-electron oxidation of the precursor peptide (MbnA) to install an oxazolone and an adjacent thioamide, the characteristic methanobactin bidentate copper ligands. MbnB and MbnC homologs are encoded together and separately in many bacterial genomes, suggesting functions beyond their roles in methanobactin biosynthesis.
Heather Pinkett Lab
Effects of lipid environment on the conformational changes of an ABC importer
Austin J Rice, Fracnes JD Alvarez, Amy L Davidson, Heather W Pinkett
In order to shuttle substrates across the lipid bilayer, membrane proteins undergo a series of conformation changes that are influenced by protein structure, ligands, and the lipid environment. To test the effect of lipid on conformation change of the ABC transporter MolBC, EPR studies were conducted in lipids and detergents of variable composition. In both a detergent and lipid environment, MolBC underwent the same general conformation changes as detected by site-directed EPR spectroscopy. However, differences in activity and the details of the EPR analysis indicate conformational rigidity that is dependent on the lipid environment. From these observations, we conclude that native-like lipid mixtures provide the transporter with greater activity and conformational flexibility as well as technical advantages such as reconstitution efficiency and protein stability.
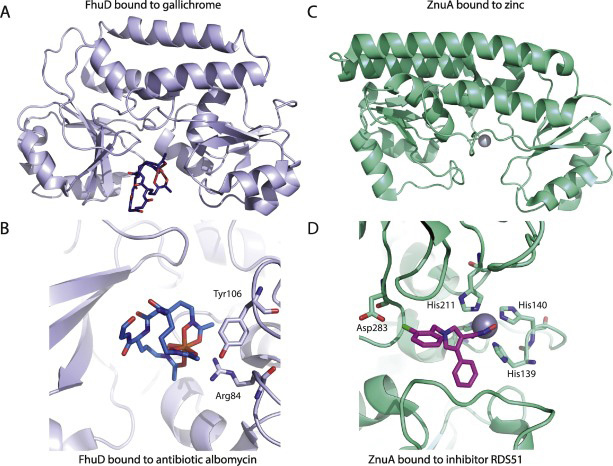
Selective substrate uptake: The role of ATP-binding cassette (ABC) importers in pathogenesis
Kari J Tanaka, Saemee Song, Kevin Mason, Heather W Pinkett
The uptake of nutrients, including metals, amino acids and peptides are required for many biological processes. Pathogenic bacteria scavenge these essential nutrients from microenvironments to survive within the host. Pathogens must utilize a myriad of mechanisms to acquire these essential nutrients from the host while mediating the effects of toxicity. Bacteria utilize several transport proteins, including ATP-binding cassette (ABC) transporters to import and expel substrates. ABC transporters, conserved across all organisms, are powered by the energy from ATP to move substrates across cellular membranes. In this review, we will focus on nutrient uptake, the role of ABC importers at the host–pathogen interface, and explore emerging therapies to combat pathogenesis.
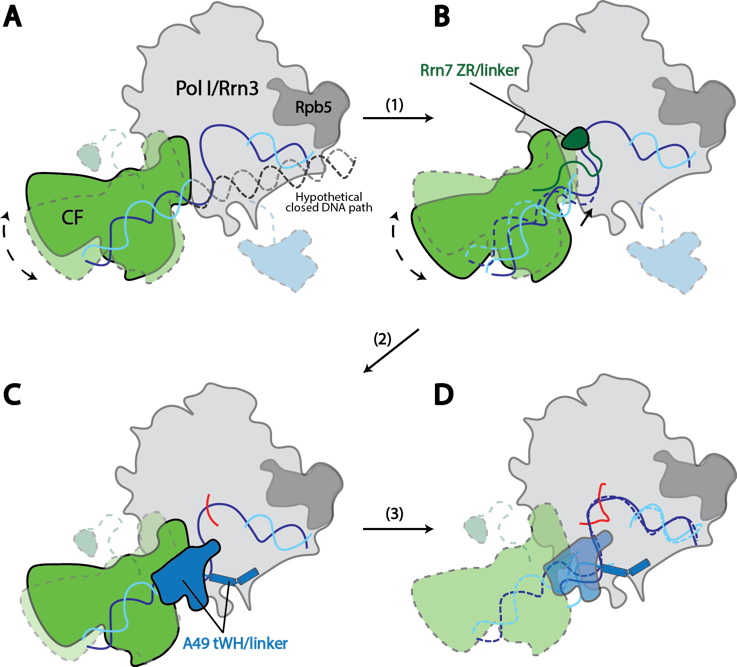
Yuan He Lab
Structural Mechanism of ATP-independent Transcription Initiation by RNA Polymerase I
Yan Han, Chunli Yan, Thi Hoang Duong Nguyen, Ashleigh J. Jackobel, Ivaylo Ivanov, Bruce Knutson, Yuan He
Transcription initiation by RNA Polymerase I (Pol I) depends on the Core Factor (CF) complex to recognize the upstream promoter and assemble into a Pre-Initiation Complex (PIC). Here, we solve a structure of Saccharomyces cerevisiae Pol I-CF-DNA to 3.8Å resolution using single-particle cryo-electron microscopy (cryo-EM). The structure reveals a bipartite architecture of Core Factor and its recognition of the promoter from -27 to -16. Core Factor’s intrinsic mobility correlates well with different conformational states of the Pol I cleft, in addition to the stabilization of either the Rrn7 N-terminal domain near Pol I wall or the tandem winged helix domain of A49 at a partially overlapping location. Comparison of the three states in this study with the Pol II system suggests that a ratchet motion of the Core Factor-DNA sub-complex upstream facilitates promoter melting in an ATP-independent manner, distinct from a DNA translocase actively threading the downstream DNA in the Pol II PIC.
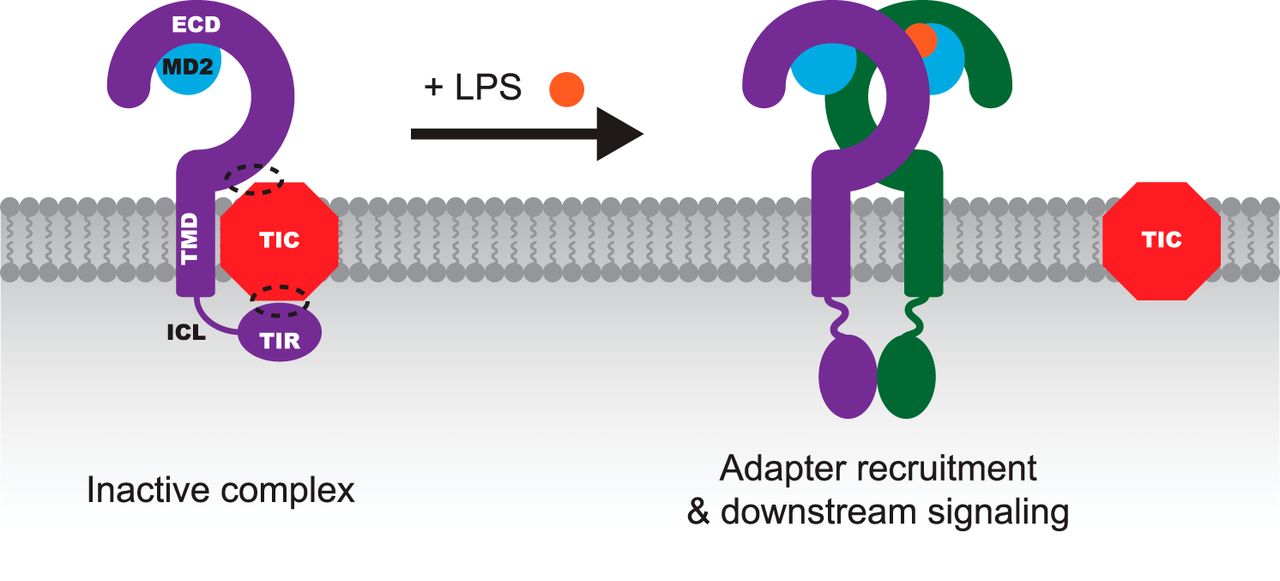
Joshua Leonard Lab
Contributions of Unique Intracellular Domains to Switchlike Biosensing by Toll-like Receptor 4
Nichole M. Daringer, Kelly A. Schwarz and Joshua N. Leonard
Toll-like receptors (TLRs) mediate immune recognition of both microbial infections and tissue damage. Aberrant TLR signaling promotes disease; thus, understanding the regulation of TLR signaling is of medical relevance. Although downstream mediators of TLR signaling have been identified, the detailed mechanism by which ligand binding-mediated dimerization induces downstream signaling remains poorly understood. Here, we investigate this question for TLR4, which mediates responsiveness to bacterial LPS and drives inflammatory disease. TLR4 exhibits structural and functional features that are unique among TLRs, including responsiveness to a wide variety of ligands. However, the connection between these structural features and the regulation of signaling is not clear. Here, we investigated how the unique intracellular structures of TLR4 contribute to receptor signaling. Key conclusions include the following. 1) The unique intracellular linker of TLR4 is important for achieving LPS-inducible signaling via Toll/IL-1 receptor (TIR) domain-containing adapter-inducing interferon-β (TRIF) but less so for signaling via myeloid differentiation primary response 88 (MyD88). 2) Membrane-bound TLR4 TIR domains were sufficient to induce signaling. However, introducing long, flexible intracellular linkers neither induced constitutive signaling nor ablated LPS-inducible signaling. Thus, the initiation of TLR4 signaling is regulated by a mechanism that does not require tight geometric constraints. Together, these observations necessitate refining the model of TLR4 signal initiation. We hypothesize that TLR4 may interact with an inhibitory partner in the absence of ligand, via both TIR and extracellular domains of TLR4. In this speculative model, ligand binding induces dissociation of the inhibitory partner, triggering spontaneous, switchlike TIR domain homodimerization to initiate downstream signaling.
NEIL Kelleher Lab
Total Kinetic Analysis Reveals How Combinatorial Methylation Patterns are Established on Lysines 27 and 36 of Histone H3
, , , , , , and
The great DNA sequencing technology leap in recent years has made it possible to sequence more and more genome from individual cancer patients. The rationale behind these great efforts is that the recurring mutations, constantly showing up in many cancer patients, are the "driver" mutations likely to cause cancer whereas rare mutations are just "passenger" mutations and therefore not critical. Surprisingly, mutations in genes encoding histone modifying enzymes turn out to be such "driver" mutations. These enzymes can "write" and "erase" some small tags (methylation, acetylation, phosphorylation, etc.) in the tail of histones, which pack our DNA into the form of chromatin. The complex combinations of these different modifications and many different outcomes dictated by various "readers" have been the central part of "histone code" theory.
Importantly, research suggests that abnormal chromatin landscape caused by these recurring mutations is the molecular culprit leading to cancer since histone modification can influence how cells use the information stored in DNA. Because this process is not involved in the sequence of DNA itself, it is referred as epigenetic. In contrast to the genetic mutations, histone modifications are reversible. Therefore, we can restore the chromatin landscape from disease state to normal state if we can manipulate the "writing" and "erasing" speed of deregulated histone modifying enzymes.
To study how aberrant histone modifications are established in multiple myeloma patients with overproduction of histone methyltransferase (MMSET) as result of chromosome translocation, we developed an integrated approach Mass Spectrometry-based Measurement and Modeling of Histone Methylation Kinetics ("M4K") by combining stable isotopes, quantitative mass spectrometry and computational modeling to determine the full matrix of effective rate constants for two methylation sites at K27 and K36 of histone H3. M4K revealed that when H3K36 or H3K27 is dimethylated, then rates of further methylation on the other site are reduced precipitously, which explains why both H3K27 and H3K36 methylation patterns are affected in multiple myeloma (bi-directional antagonism model). Another surprise discovery is the increase methylation turnover at H3K27 when MMSET is overexpressed. Most significantly, we can use the kinetic model obtained by M4K to predict how much inhibition is required to restore the normal epigenetic state in these cancer cells.
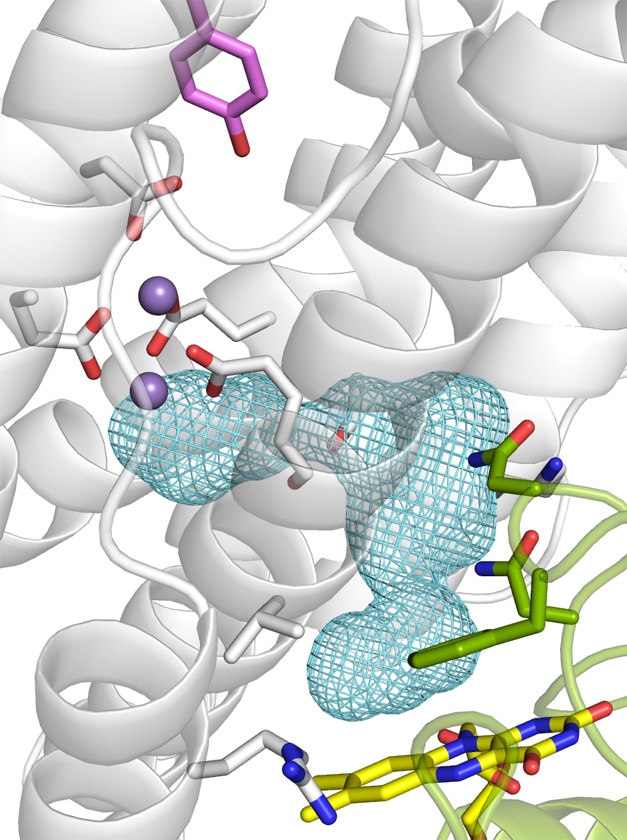
Amy Rosenzweig Lab
Structural Basis for Activation of Class Ib Ribonucleotide Reductase
Amie K. Boal, Joseph A. Cotruvo, Jr., JoAnne Stubbe, and Amy C. Rosenzweig
The class Ib ribonucleotide reductase of Escherichia coli can initiate reduction of nucleotides to deoxynucleotides with either a MnIII2-tyrosyl radical (Y•) or a FeIII2-Y• cofactor in the NrdF subunit. Whereas FeIII2-Y• can self-assembleStructural basis for activation of class Ib ribonucleotide reductase from FeII2-NrdF and O2, activation of MnII2-NrdF requires a reducedflavoprotein, NrdI,proposed to form the oxidant for cofactor assembly by reduction of O2. The crystal structures reported here of E. coli MnII2-NrdF and FeII2-NrdF reveal different coordination environments, suggesting distinct initial binding sites for the oxidants during cofactor activation. In the structures of MnII2-NrdF in complex with reduced and oxidized NrdI, a continuous channel connects the NrdI flavin cofactor to the NrdF MnII2 active site. Crystallographic detection of a putative peroxide in this channel supports the proposed mechanism of MnIII2-Y• cofactor assembly. (PDF)
Brian Hoffman Lab
Photoinitiated Singlet and Triplet Electron Transfer Across a Re-Designed [Myoglobin, Cytochrome b5] Interface
Judith M. Nocek, Amanda K. Knutson, Peng Xiong, Nadia Petlakh Co, and Brian M. Hoffman
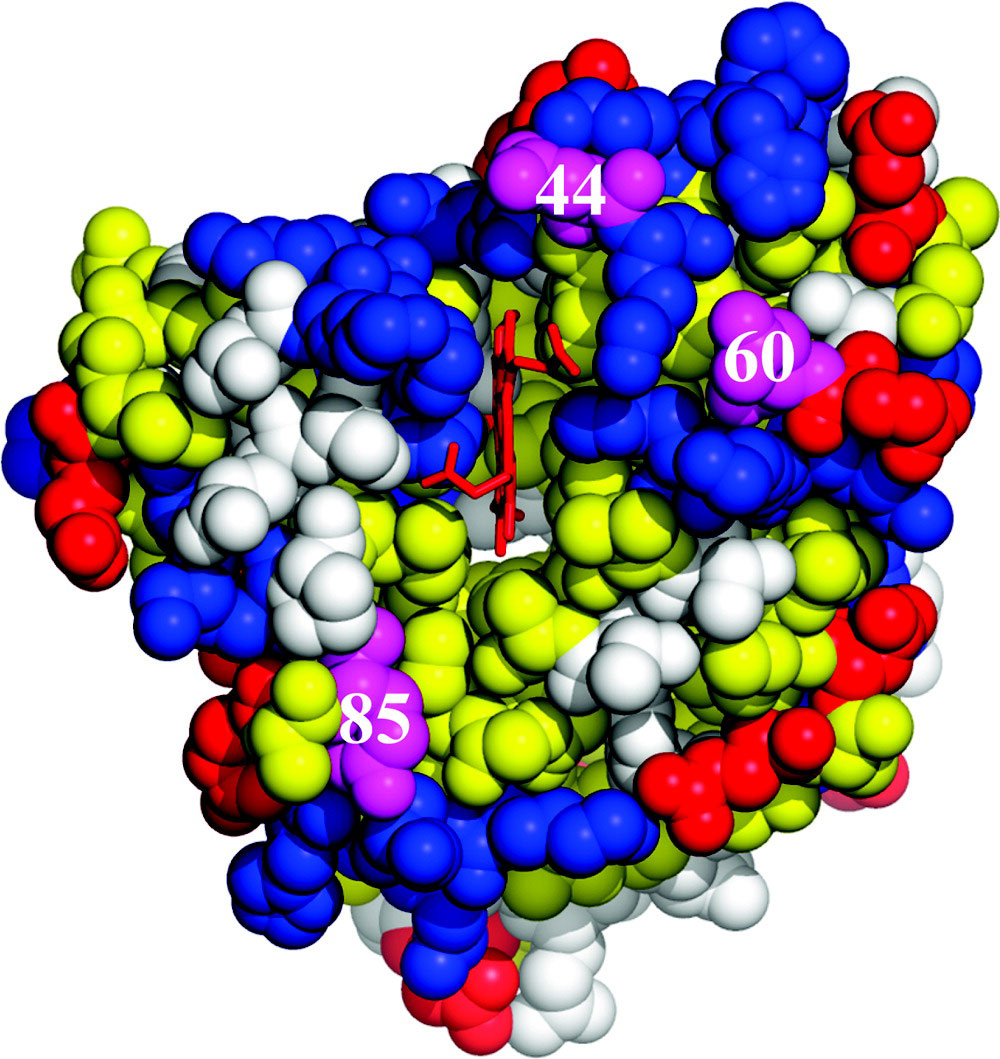
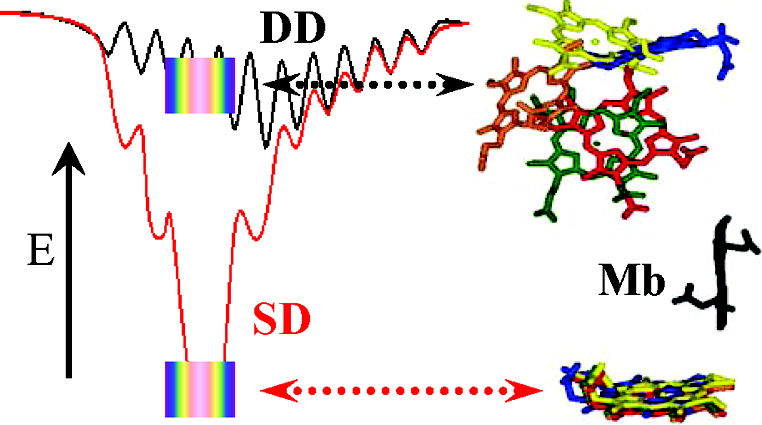
Abstract: We describe a strategy by which reactive binding of a weakly-bound, ‘dynamically docked (DD)’ complex without a known structure can be strengthened electrostatically through optimized placement of surface charges, and discuss its use in modulating complex formation between Photoinitiated Singlet and Triplet Electron Transfer Across a Re-Designed [Myoglobin, Cytochrome b5] Interfacemyoglobin (Mb) and cytochrome b5 (b5). The strategy employs paired Brownian Dynamics (BD) simulations, one which monitors overall binding, the other reactive binding, to examine [X g K] mutations on the surface of the partners, with a focus on single and multiple [D/E g K] charge reversal mutations. This procedure has been applied to the [Mb, b5] complex, indicating mutations of Mb residues D44, D60 and E85 to be the most promising, with combinations of these showing a nonlinear enhancement of reactive binding. A novel method of displaying BD profiles shows that the ‘hits’ of b5 on the surfaces of Mb(WT), Mb(D44K/D60K), and Mb(D44K/D60K/E85K) progressively coalesce into two ‘clusters’: a ‘diffuse’ cluster of hits that are distributed over the Mb surface and have negligible electrostatic binding energy; a ‘reactive’ cluster of hits with considerable stability that are localized near its heme edge, with short Fe-Fe distances favorable to electron transfer (ET). Thus binding and reactivity progressively become correlated by the mutations. This finding fits well with recent proposals that complex formation is a two-step process, proceeding through the formation of a weakly-bound encounter complex (‘diffuse cluster’) to a well-defined bound complex (‘reactive cluster’). The design procedure has been tested through measurements of photoinitiated ET between the Zn-substituted forms of Mb(WT), Mb(D44K/D60K) and Mb(D44K/D60K/E85K) and Fe3+b5. Both mutants convert the complex from the DD regime exhibited by Mb(WT), in which the transient complex is in fast kinetic exchange with its partners, koff >> ket, to the slow-exchange regime, ket >> koff, and both mutants exhibit rapid intracomplex ET from the triplet excited state to Fe3+b5 (rate constant, ket ~ 106 s-1). The affinity constants of the mutant Mbs cannot be derived through conventional analysis procedures because intracomplex singlet ET quenching causes the triplet-ground absorbance difference to progressively decrease during a titration, but this effect has been incorporated into a new procedure for computing binding constants. Most importantly, this is the first evidence for photo-induced singlet ET across a protein-protein interface.
Electrostatic Redesign of the [Myoglobin,Cytochrome b5] Interface To Create a Well-Defined Docked Complex with Rapid Interprotein Electron Transfer
Peng Xiong, Judith M. Nocek, Amanda K. K. Griffin, Jingyun Wang, and Brian M. Hoffman
Cyt b5 is the electron-carrier ‘repair’ protein that reduces met-Mb and met-Hb to their O2-carrying ferroheme forms. Studies of electron transfer (ET) between Mb and cyt b5 revealed that they react on a “Dynamic Docking” (DD) energy landscape on which binding and reactivity are uncoupled: binding is weak and involves an ensemble of nearly isoenergetic configurations, only a few of which are reactive; those few contribute negligibly to binding. We set the task of redesigning the surface of Mb so that its reaction with cyt b5 instead would occur on a conventional ‘simple docking’ (SD) energy landscape, on which a complex exhibits a well-defined (set of) reactive binding configuration(s), with binding and reactivity thus no longer being decoupled. We prepared a myoglobin (Mb) triple mutant (D44K/D60K/E85K; Mb(+6)) substituted with Zn-deuteroporphyrin, and monitored cytochrome b5 (cyt b5) binding and electron transfer (ET) quenching of the 3ZnMb(+6) triplet state. In contrast, to Mb(WT), the three charge-reversals around the ‘front-face’ heme edge of Mb(+6) have directed cyt b5 to a surface area of Mb adjacent to its heme, created a well-defined, most-stable structure that supports good ET pathways, and apparently coupled binding and ET: both Ka and ket are increased by the same factor of ~ 2×102, creating a complex that exhibits a large ET rate constant, ket = 106 1s−1, and is in slow exchange (koff ≪ ket). In short, these mutations indeed appear to have created the sought-for conversion from DD to simple docking (SD) energy landscapes.
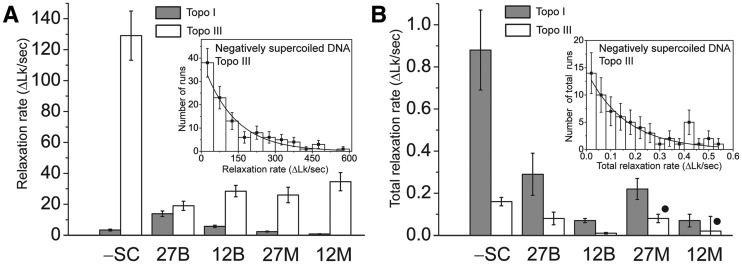
JOhn Marko and Alfonso Mondragón LABS
Bacterial topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways
Ksenia Terekhova, Kathryn Gunn, John Marko, Alfonso Mondragón
Escherichia coli topoisomerases I and III (Topo I and Topo III) relax negatively supercoiled DNA and also catenate/decatenate DNA molecules containing single-stranded DNA regions. Although these enzymes share the same mechanism of action and have similar structures, they participate in different cellular processes. In bulk experiments Topo I is more efficient at DNA relaxation, whereas Topo III is more efficient at catenation/decatenation, probably reflecting their differing cellular roles. To examine the differences in the mechanism of these two related type IA topoisomerases, single-molecule relaxation studies were conducted on several DNA substrates: negatively supercoiled DNA, positively supercoiled DNA with a mismatch and positively supercoiled DNA with a bulge. The experiments show differences in the way the two proteins work at the single-molecule level, while also recovering observations from the bulk experiments. Overall, Topo III relaxes DNA efficiently in fast processive runs, but with long pauses before relaxation runs, whereas Topo I relaxes DNA in slow processive runs but with short pauses before runs. The combination of these properties results in Topo I having an overall faster total relaxation rate, even though the relaxation rate during a run for Topo III is much faster.